Weaver DF (2023) Alzheimer's disease as an innate autoimmune disease (AD2): A new molecular paradigm. Alzheimers Dement 19, 1086-1098.
Recently, Alzheimer’s disease (AD) was officially nominated to the ever-growing list of what now constitutes, according to The National Institutes of Health, more than 80 “autoimmune” diseases, covering everything from Stiff-Person Syndrome to Diabetes Type 1 (https://www.niehs.nih.gov/health/topics/conditions/autoimmune/index.cfm). The thoughts behind Dr. Donald F. Weaver’s well-designed and informative “Alzheimer’s disease as an autoimmune disease” get fairly involved, in fact quite intricate. Generally, Dr. Weaver’s theory is explained as “a brain-centric disorder of innate immunity involving concurrent autoimmune and autoinflammatory mechanisms”. Although the theory still includes Aβ as an important molecular player, it rejects the “amyloid misfolding hypothesis” per se.
So far, so good.
Weaver continues to persuasively simplify his case for AD as an autoimmune disease:
“We believe that beta-amyloid is not an abnormally produced protein, but rather is a normally occurring molecule that is part of the brain’s immune system. It is supposed to be there. When brain trauma occurs or when bacteria are present in the brain, beta-amyloid is a key contributor to the brain’s comprehensive immune response. And this is where the problem begins.
Because of striking similarities between the fat molecules that make up both the membranes of bacteria and the membranes of brain cells, beta-amyloid cannot tell the difference between invading bacteria and host brain cells, and mistakenly attacks the very brain cells it is supposed to be protecting.
This leads to a chronic, progressive loss of brain cell function, which ultimately culminates in dementia — all because our body’s immune system cannot differentiate between bacteria and brain cells. When regarded as a misdirected attack by the brain’s immune system on the very organ it is supposed to be defending, Alzheimer’s disease emerges as an autoimmune disease.”
What could go wrong with that? Well, nothing, except perhaps for the present thoughts regarding autoimmunity weighed against other evidence.
Introduction
 Paul Ehrlich (1854-1915)
Paul Ehrlich (1854-1915)
The organism possesses certain contrivances by means of which the immunity reaction, so easily produced by all kinds of cells is prevented from acting against the organism’s own elements and so giving rise to auto toxins …so that one might be justified in speaking of a “horror Autotoxicus” [the horror of self-toxicity] of the organism. These contrivances are naturally of the highest importance for the existence of the individual.
˗ Paul Ehrlich
Ehrlich P, Morgenroth J (1901) “Studies on Hemolysins” Berliner kin Wochenschrift. No 10. Reprinted in Collected studies on Immunity (1906).
Thus, while investigating autoimmunity, the great German bacteriologist and immunologist Paul Ehrlich (1854-1915) rejected the then new hypothesis of autoimmunity: that an organism's immune system could attack the body’s own tissue, coining it "horror autotoxicus", literally meaning, the horror of self-toxicity. He used this term to describe the body's innate aversion to immunological self-destruction, insisting that horror autotoxicus itself would prevent such self-toxicity in a living organism. This wasn’t just any scientist. Ehrlich was to immunology what Einstein was to relativity, and by 1908 he shared a Nobel Prize in Medicine for his work on the immune system. Instrumental in immunology’s very basics, he theorized the very existence of antibodies: proteins with shapes that bind to the proteins that stick out of pathogens, in lock and key fashion. He also was responsible for the first treatment for diphtheria, Gram staining methods to categorize bacteria, and the development of chemotherapy as we know it. Because of the dualism of specificity of pathogen and host response, the development of modern bacteriology gave rise to the birth of immunology. Thus, Robert Koch paved the way for immunology put forward by his disciple Paul Ehrlich. But Paul Ehrlich had conducted animal experiments which led him to firmly come out against autoimmunity. Animals injected with different species’ blood formed antibodies against such foreign cells, while those injected with the same species did not form disease-causing autoantibodies. To Ehrlich this meant that the immune system would not do something as counterproductive as damaging its own healthy cells, and thus he could not subscribe to this new concept of autoimmunity.
Yehuda Shoenfeld’s Infection and Autoimmunity (2005) long ago came to the conclusion that all autoimmune diseases were infectious until proven otherwise. Infections themselves have long been considered important environmental triggers of autoimmunity, its onset and its severity. But the difference between “trigger” and “cause” sometimes wore thin. And when Shoenfeld revisited the subject in his second edition (2015) of Infection and Autoimmunity, he devotes an entire chapter to the concept of Mycobacteria and Autoimmunity, with 13 annotated references [1-14].
The association between immune response against mycobacterial infections and autoimmune disease has long been suspected, including systemic lupus erythematosus (SLE), Type 1 diabetes mellitus, multiple sclerosis (MS), inflammatory bowel disease (IBD), microscopic polyangiitis, and sarcoidosis (SA). Relatively recently Stiff-Person Syndrome, felt to be autoimmune, and which causes rigidity and spasms in the trunk and limbs, has been added to this list as an autoimmune and neurological disorder.
In his book Autoimmune Aspects of Lung Disease (1998), Isenberg maintained that not only is tuberculosis itself accompanied by a spectrum of autoantibodies like that seen in autoimmune disease, but that the mycobacteria in general, such as tuberculosis, leprosy, Mycobacterium avium and even the TB vaccination Bacille-Calmatte-Guérin can all be accompanied by syndromes resembling diseases conventionally labelled as “autoimmune” [15]. For example, reactivity to the mycobacterial 65 kDa heat shock protein (hsp 65) has been implicated in the pathogenesis of adjuvant arthritis in the rat, and may be involved in the pathogenesis of rheumatoid arthritis or other autoimmune diseases in humans. And that because similar phenomena can be seen in even non-mycobacterial infections (including Lyme disease and syphilis), “the cause of ‘autoimmune’ diseases must be questioned.” No one is challenging that infections such as tuberculosis can use their long arm of immune phenomena to damage cells in the body. Recent experimental evidence allows for an immunological basis for mycobacterial AD-like conditions. Hernández-Pando’s group [16], in late 2020, was able to affect long and short-term memory deficits without having to introduce experimental infection directly into the brain at all. Pando’s group, through the long reach of severe tubercular cell-mediated immunological reactions alone, caused significant amounts of damage to the memory centers in the brain, its neurons and its synapses simply thru initiating a focus of pulmonary tuberculosis in the lungs. They also noted a marked increased synthesis of both inflammatory and anti-inflammatory cytokines in discrete brain areas such as the hypothalamus, the hippocampal formation and cerebellum accompanied by substantial changes in the synthesis of neurotransmitters.
The Rise and Fall of “Autoimmune” Whipple’s
It is not easy to differentiate an “autoimmune” from an infectious disorder. An instructive case in point is the saga of Whipple’s disease. At one time, Whipple’s, a rare cause of chronic diarrhea and abdominal pain, easily confused with inflammatory bowel disease such as Crohn’s and Ulcerative colitis, was also thought to be an autoimmune disease [17]. Until the early 1960s, the disease was considered a uniformly fatal and untreatable primary disorder of fat metabolism. But the cause of Whipple’s disease turned out to be anything but “autoimmune” and its true origin turned out to be a pathogen related to what Whipple first described in 1907 in a paper in the now-defunct Bulletin of Johns Hopkins Hospital [18]. At that time Whipple reported the isolation of a “Rod-shaped organisms in silver-stained gland tissue, closely resembling the tubercle bacillus”. But it took 100 years, in 2003, to prove that he was right. Using novel diagnostic methods on stored tissue samples from Whipple’s original patient, investigators found T. whipplei, a microbial distant relative of Mycobacterium avium (Fowl tuberculosis), and Mycobacterium paratuberculosis [19]. This validated another study wherein as early as 1992, Rook and Stanford insisted that “autoimmune” disease, which at that time included Whipple’s disease, was tied to “conventional mycobacterioses’, such as tuberculosis [20].
On “Stiff” Persons
Then, lately, was added Stiff-Person Syndrome (SPS), formerly Stiff-Man Syndrome, to the ever-burgeoning list of “Autoimmunity”, although the exact cause of SPS is admittedly unknown. But, at the end of the day, diagnosing a person as having “Stiff-Person Syndrome” is nothing but a rudimentary, descriptive, observational diagnosis, bolstered to a limited extent by the lab findings of antibodies, mostly directed against the 65-kD form of Glutamic Acid Decarboxylase (GAD65) enzyme, which might or might not have anything to do with the condition. Since most patients with “classical” SPS have anti-GAD antibodies, it is currently believed that the possible pathological mechanism of SPS is the inhibition of GABA by these anti-GAD antibodies, leading to overexcitation of motor neurons, resulting in continuous muscle rigidity and painful spasms. In actuality, the pancreas needs the enzyme glutamic acid decarboxylase (GAD) to function normally. Antibodies that target this enzyme are called GAD antibodies. This does not automatically make them “autoimmune”. And certainly, not all patients with GAD65 antibody in the CSF and the serum will show SPS.
Furthermore, long before antibodies directed against GAD65 were considered causative of the then “Stiff-man syndrome”, and autoimmune Diabetes Mellitus type 1, it was thought that circulating antibodies against Mycobacterial heat shock protein 65 (hsp65), instigated by stressful condition such as heat, were the true cause of many clinical diseases including the autoimmune diseases Crohn’s disease, lupus erythematosus, multiple sclerosis, diabetes, and others. Certainly, elevated levels of immune response targeting both GAD 65 and such heat shock protein were both elevated in Diabetes type 1. Then it became obvious that GAD65 and Hsp 65 have substantially similar amino acid sequences [21]. The linkage of what used to be called Stiff-Man Syndrome (SMS) to Diabetes type 1 is undeniable. Insulin-dependent Type 1 diabetes mellitus, also felt to be caused by autoimmune destruction, destroys the insulin-producing beta cells of the pancreas. While most patients with the classical form of ‘Sick-Person Syndrome’ (SPS) have auto-antibodies to GAD, almost all patients positive for this antibody are also positive for islet cell cytoplasmic antibodies and a significant proportion have insulin dependent diabetes.
The results described by Elias et al. indicate that a beta-cell target antigen in non-obese diabetic (NOD/Lt) mice is a molecule cross-reactive with the 65-kDa heat shock protein (hsp65) of Mycobacterium tuberculosis. Moreover, this tubercular HSP65 antigen could be used either to induce diabetes, or to vaccinate against diabetes, depending on the form of its administration to pre-diabetic NOD/Lt mice [22]. A specific role of mycobacterial HSPs in the etiology of autoimmune disease is further suggested by the development of adjuvant arthritis in rats following a single injection of an extract of heat killed M. tuberculosis in Freund's complete adjuvant. Injected rats develop T cells specifically reacting with mycobacterial HSP and such T cells can induce the arthritis when transferred to irradiated rats which have never been exposed to M. tuberculosis. By means of specificity analysis of cloned T cells, a 65 kD heat shock protein of mycobacteria was identified as crucial to instigating this autoimmune arthritis. In fact, evidence accumulated suggesting that the same hsp65 might be crucial in human chronic arthritis in general [23]. Moreover, in as much as one-third of patients, SPS has a focal presentation, limited to rigidity and painful spasms of the trunk or to one limb, (the so-called “focal” SPS). In such cases, which are many, as low as 15% (15-61%) are GAD65 antibody positive. Even classical SPS patients can have, in some studies, only 60% GAD Ab detection, and all of the rest are seronegative or associated with other neural antibodies (amphiphysin, glycine receptor). Add to this the fact that other not directly related neurologic disease can also throw off the very same high titers of these same GAD antibodies, including so-called “autoimmune” cerebellitis, brain stem encephalitis, seizure disorders, and other myelopathies [now neatly swept under the rug as “GAD antibody-spectrum disorders (GAD-SD)”], and one can easily ascertain why Stiff-person syndrome remains a clinical diagnoses, difficult because it is largely based on clinical symptoms, most of which just happens to mimic, down to its last sign and symptom other older neuromuscular disorders.
Preconceived Notions/Faulty Screening
But again, there are big time problems with the way experimental protocols are being performed to do the all-important rule-outs of infectious disease screening before labeling them “autoimmune”. Let’s take for example, the recently initiated “GAD antibody-spectrum disorder (GAD-SD)” called “autoimmune encephalitis”, presently recognized in patients with autoimmunity related to the 65-kDa isoform of glutamic acid decarboxylase (GAD65) antibodies, even if patients with meningoencephalitis associated with those antibodies have been rarely identified. In the 2023 study “Meningoencephalitis associated with GAD65 autoimmunity”, Kuang et al. [24] chose, under their infectious disease screening protocols, to do a mere CSF (cerebrospinal fluid) acid-fast (tubercular) stain to detect and thus rule out central nervous system tuberculosis. Unfortunately, such inadequate staining is not an uncommon practice in studies ruling out mycobacterial infection when primarily seeking “autoimmunity”. In fact it is the norm, and as a result [25], potentially life-saving antibiotics are removed, and IV methylprednisolone, alone, which can spread infection, begun.
Yet Stadelman et al., in 2022, concluded [26] that a CSF tubercular acid-fast bacillus (AFB) smear has poor sensitivity in most settings, and that many studies demonstrated low (10–15%) sensitivity for CSF AFB smear, and that therefore a significant number of cases are missed using this technique, often with faulty diagnosis and fatal consequences [27]. The problem is it happens all the time: in the lab, in the hospital… Stadelman et al. instead concluded that at present, the combination of GeneXpert MTB/Rif Ultra (a molecular, rapid diagnostic test that simultaneously detects M. tuberculosis complex and rifampicin resistance] and culture present the best chance to diagnose TB meningitis in most settings, although each of these can discover cases that the other test misses. Such is the rigor with which present-day diagnosis should and must be pursued, and even a combination of these tests is far from fool-proof.
Alzheimer’s Amyloid: Nothing “Autoimmune” about it
In their landmark article “Can infections cause Alzheimer’s disease?”, Mawanda and Wallace came to the conclusion that “amyloidopathy – a condition characterized by elevated levels of serum amyloid and by amyloid deposition and aggregation in tissues – is a frequent occurrence in several acute and chronic systemic inflammatory conditions, especially chronic infections like tuberculosis and leprosy.” [28,29]. And rifampin, a first line drug against TB has unquestionably proved to be protective against the progress of AD, the neurotoxicity of its amyloid beta protein, and its tau oligomers [30-32].
Anti-tubercular therapy led to resolution of enhancing lesions in the brain and abatement of memory deficits [33]. And a prime example of non-tubercular mycobacteria (NTM) involvement includes the often-pathogenic Mycobacterium avium complex. A recent 2020 publication found that prominent among the complaints given by NTM infected patients and reported by their physicians were “memory problems” [34].
From its onset, tuberculosis (Mtb) is intimately and massively tied to the production of amyloid because of its proven ability to specifically bind and exponentially elevate human serum Amyloid (SAA1). Tubercle bacilli, opsonized with the physiological and 5-fold higher concentrations of this human acute phase of infection amyloid, primes the path for the substantial SAA1-promoted entry of tubercular bacilli into macrophages that normal physiological concentrations of SAA1 would never have allowed [35]. Scientific reports have documented a significantly increased concentration of SAA in patients with tuberculosis [36-38]. Prolonged or cyclic elevation of circulating SAA typically precedes the deposition of AA amyloid. When chronic elevations of SAA are from infection though, obviously it is the chronic infections which will lead to greater amyloid deposition.
Researcher and neuropathologist Phillip Schwartz, a pioneer in neurodegenerative amyloid disease, and the innovator of the Thioflavin dye used to detect it, essentially pre-dated what Mawanda and Wallace concluded in “Can infections cause Alzheimer’s disease?” Schwartz:
Chronic infections, and particularly tuberculosis, have heretofore been regarded as the most frequent and therefore the most important cause of systemic amyloid degeneration. This fact induced us to look for sequels of pulmonary tuberculosis in around 150 cases of senile and presenile [Alzheimer’s] amyloidosis. Postmortem roentgen photography, as well as careful macroscopic and histologic examination of the lungs, disclosed typical calcifications of healed intra-pulmonary or lymphoglandular tuberculous infiltrates in almost every instance of this series, and quite often typical cicatrices of old lymphonodular bronchial lesions. Doubtless, the great majority of our senile and presenile patients displaying cerebral amyloidosis, suffered from pulmonary tuberculosis sometimes before symptoms of senile deterioration arose [39].
As if the ability to sharply elevate and then bind serum amyloid five-fold in its acute phase, mainly through the liver, were not enough, the tubercular bacilli itself is a major manufacturer of amyloid, churning this out courtesy of its major amyloidogenic substrate ESAT-6 secretion system-1, also called ESX-1. The identification of ESX-1 as a major virulence determinant of Mycobacterium tuberculosis has been a major discovery in the history of tuberculosis research. The ESAT-6 gene is located within the RD1 region (region of differences), a chromosomal region present mainly in pathogenic mycobacteria and deleted from all M. bovis BCG strains examined so far. It was the discovery that tubercular RD1 mediates the induction of macrophage death which established its critical importance in the virulence of M. tuberculosis pathogenicity. But there is more, much more.
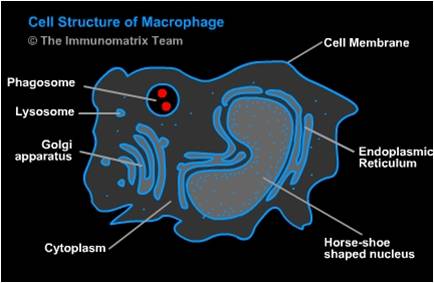
Fig. 1. Cell Structure of Macrophage [(Salvador Miranda: blogs.hightechhigh.org/smiranda47/files/2013/03/macrophagecelldiag.jpg). In https://openwetware.org/wiki/Macrophages_and_Their_Interactions_with_Engineered_Tissues,_by_Veronica_Murray
After the phagosome “matures”, the body’s macrophage does what it does with all intracellular intruders, it attempts by to kill the trespasser through fusion with multiple lysosomes and their digestive enzymes.
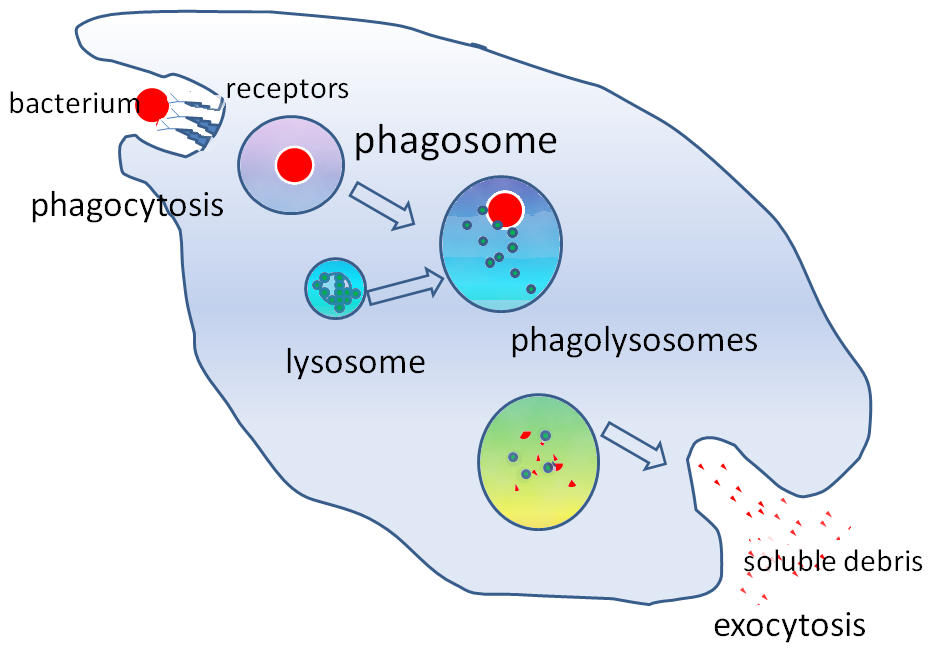
Fig. 2. Lysosome/phagosome fusion. From https://en.wikipedia.org/wiki/Phagosome
At that point it should be game over for the number one infectious killer in the world, except that early in lysosome fusion virulent M. tuberculosis, through the eons has learned to escape enzymatic destruction by moving from the confines of the membrane bound phagosome and winding up in the cytosol or aqueous component of the macrophage’s cytoplasm, where it continues to replicate. This “Great Escape”, has been perhaps the costliest in terms of human life in the history of infectious death. And it is brought to us courtesy of the virulent ESX-1/ESAT-6 in the region of difference, and its ability to cause phagosomal membrane disruption. If a mycobacterium such as M. smegmatis is unable to rupture the phagosome membrane, it will face enzymatic death upon lysosomal fusion.
Furthermore, this same ability of tubercular ESAT-6 to punch tiny holes (pores-forming) through membranes leading to escape, is also responsible for the formation of pore forming small oligomers, composed from amyloidogenic intermediate soluble proteins created by tubercular ESAT-6, and not found in mutants without it [40]. Such toxic soluble intermediates, often referred to as amyloid-β oligomers (AβOs), the most toxic and pathogenic form of Aβ, are believed, in turn, to be the precursor of insoluble amyloid [41], the accumulation of which is associated with not only AD, but Parkinson’s disease, so-called Type 2 diabetes, and a host of other diseases. Such aggregation of amyloid, although it wreaks havoc on the human system, has been looked upon appropriately as merely a detoxification mechanism in response to the noxious intermediate substances churned out by ESAT-6 [42]. Indeed once inside the macrophage, it is the ESX-1/ESAT-6 secretion apparatus of MTB which not only damages the phagosome leading to mycobacterial escape, but eventually induces macrophage lysis, and NLRP3 inflammasome release.
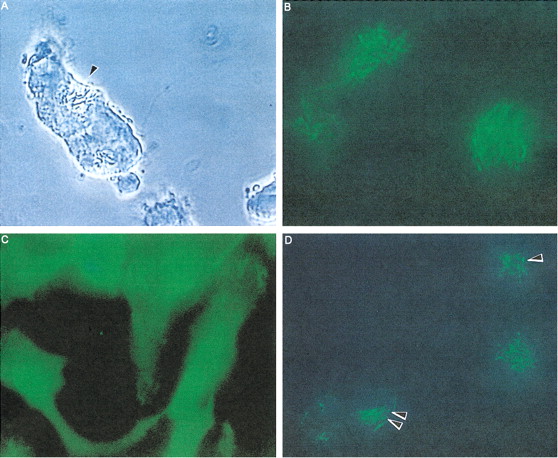
Fig. 3. Fusion of Mycobacterium smegmatis and Mycobacterium avium phagosomes. A) Time lapse video microscopy showing fusion of M. avium and M. smegmatis vacuoles after 2 days of coinfection. B) Acridine orange staining of M. smegmatis in RAW 264.7 macrophages after 2 days of infection. Bacteria are observed incorporating the dye. C) M. avium (48 h)–infected macrophages stained with acridine orange. No bacterium incorporating the dye is seen, indicating that M. avium is in a nonacidic environment. D) RAW 264.7 macrophages infected with M. avium for 24 h and subsequently with M. smegmatis for 48 h. The figure shows M. smegmatis (long rods) and M. avium (short rods; arrowheads) stained with acridine orange, indicating that both are in acidic environment. From Broxmeyer L, Sosnowska D, Miltner E, Chacón O, Wagner D, McGarvey J, Barletta RG, Bermudez LE. Killing of Mycobacterium avium and Mycobacterium tuberculosis by a mycobacteriophage delivered by a nonvirulent mycobacterium: a model for phage therapy of intracellular bacterial pathogens. J Infect Dis. 2002 Oct 15;186(8):1155-60, with permission.
Quite recently (2022), the link between autoimmunity and tuberculosis has once again been investigated [43]. Diagnosing a disease as autoimmune has consequences, some of them potentially quite harmful. For example, corticosteroids and other immunosuppressives are sometimes used as monotherapy or in combination with other drugs for “Stiff Person Syndrome” (SPS) patients. These are the same corticosteroids and immunosuppressives that are contraindicated and can even increase the pathogenicity of certain chronic diseases, including the mycobacterial disease which more than a few investigators have implicated in neuromuscular disorders, diabetes and AD [44].
Notice: The author has no conflicting interests.
REFERENCES
[1] Dubaniewicz A (2015) Mycobacteria and Autoimmunity. In Infection and Autoimmunity, Shoenfeld Y, Agmon-Levin N, Rose NR, eds. Elsevier, pp. 551-567.
[2] Tishler M, Shoenfeld Y (1996) Anti-heat-shock protein antibodies in rheumatic and autoimmune diseases. Semin Arthritis Rheum 26, 558-53.
[3] Dubaniewicz A (2013) Microbial and human heat shock proteins as ’danger signals’ in sarcoidosis. Hum Immunol 74, 1550-1558.
[4] Dubaniewicz A, Holownia A, Kalinowski L, Wybieralska M, Dobrucki IT, Singh M (2013) Is mycobacterial heat shock protein 16 kDa, a marker of the dormant stage of Mycobacterium tuberculosis, a sarcoid antigen? Hum Immunol 74, 45-51.
[5] Dubaniewicz A (2010) Mycobacterium tuberculosis heat shock proteins and autoimmunity in sarcoidosis. Autoimmun Rev 9, 419-424.
[6] Dubaniewicz A, Kämpfer S, Singh M (2006) Serum anti-mycobacterial heat shock proteins antibodies in sarcoidosis and tuberculosis. Tuberculosis (Edinb) 86, 60-67.
[7] Dubaniewicz A, Dubaniewicz-Wybieralska M, Sternau A, Zwolska Z, Izycka-Swieszewska E, Augustynowicz-Kopec E, et al. (2006) Mycobacterium tuberculosis complex and mycobacterial heat shock proteins in lymph node tissue from patients with pulmonary sarcoidosis. J Clin Microbiol 44, 3448-3451.
[8] Dubaniewicz A, Trzonkowski P, Dubaniewicz-Wybieralska M, Dubaniewicz A, Singh M, Myśliwski A (2006) Comparative analysis of mycobacterial heat shock proteins induced apoptosis of peripheral blood mononuclear cells in sarcoidosis and tuberculosis. J Clin Immunol 26, 243-250.
[9] Cossu D, Masala S, Frau J, Mameli G, Marrosu MG, Cocco E, et al. (2014) Antigenic epitopes of MAP2694 homologous to T-cell receptor gamma-chain are highly recognized in multiple sclerosis Sardinian patients. Mol Immunol 57, 138-140.
[10] Tasneem S, islam N, Ali R (2001) Cross reactivity of SLE autoantibodies with 70 kDa heat shock proteins of Mycobacterium tuberculosis. Microbiol Immunol 45, 841-846.
[11] Sheikhi A, Nazarian M, Khadem-Al-Melleh A, et al. (2008) In-vitro effects of Mycobacterium bovis BCG-lysate and its derived heat shock proteins on cytokines secretion by blood monon unclear cells of rheumatoid arthritis patients in comparison with healthy controls. Int Immunopharmacol 8, 887-892.
[12] Komiya I, Arimura Y, Nakabayashi K, Yamada A, Osaki T, Yamaguchi H, et al. (2011) Increased concentrations of antibody against heat shock protein in patients with myeloperoxidase anti-neutrophil cytoplasmic autoantibody positive microscopic polyangiitis. Microbiol Immunol 55, 531-538.
[13] Panchapakesan J, Daglis M, Gatenby P (1992) Antibodies to 65 kDa and 70 kDa heat shock proteins in rheumatoid arthritis and systemic lupus erythematosus. Immunol Cell Biol 70, 295-300.
[14] Huszti Z, Bene L, Kovács A, Fekete B, Füst G, Romics L, Singh M, Prohászka Z (2004) Low levels of antibodies against E. coli and mycobacterial 65kDa heat shock proteins in patients with inflammatory bowel disease. Inflamm Res 53, 551-555.
[15] Isenberg DA, Spiro SG (1998) Autoimmune aspects of Lung Disease. Springer Science & Business Media, 276 pp.
[16] Lara-Espinosa JV, Santana-Martínez RA, Maldonado PD, Zetter M, Becerril-Villanueva E, Pérez-Sánchez G, et al. (2020) Experimental pulmonary tuberculosis in the absence of detectable brain infection induces neuroinflammation and behavioural abnormalities in male BALB/c mice. Int J Mol Sci 21, 9483.
[17] Weiner SR, Utsinger P (1986) Whipple disease. Semin Arthritis Rheum 15, 157-167.
[18] Whipple GH (1907) A hitherto undescribed disease characterized anatomically by deposits of fat and fatty acids in the intestinal and mesenteric lymphatic tissues. Bull Johns Hopkins Hosp 18, 382-393.
[19] Dumler JS, Baisden BL, Yardley JH, Raoult D (2003) Immunodetection of Tropheryma whipplei in intestinal tissues from Dr. Whipple's 1907 patient. N Engl J Med 348, 1411-1412.
[20] Rook GA, Stanford JL (1992) Slow bacterial infections or autoimmunity? Immunol Today 13, 160-164.
[21] Child DF, Smith CJ, Williams CP (1993) Heat shock protein and the double insult theory for the development of insulin dependent diabetes. J R Soc Med 86, 217-219. Child DF, Williams CP, Jones RP, Hudson PR, Jones M, Smith CJ (1995) Heat shock protein studies in type 1 and type 2 diabetes and human islet cell culture. Diabet Med 12, 595-599.
[22] Elias D, Markovits D, ReshefT, Van der Zee R, Cohen IR (1990) Induction and therapy of auto-immune diabetes in the non-obese diabetic (NOD/Lt) mouse by a 65 kDa heat shock protein. Proc Natl Acad Sci U S A 87, 1576-1580.
[23] Van Eden W (1990) Heat shock protein in auto-immune arthritis. A critical contribution based on the adjuvant arthritis model. APMIS 98, 383-394.
[24] Kuang Z, Baizabal-Carvallo JF, Mofatteh M, Xie S, Pan M, Ye J, Zhou L, Yang S, Wang Z, Chen Y, Li Y (2023) Meningoencephalitis associated with GAD65 autoimmunity. Front Immunol 14, 1120894.
[25] Salari M, Zaker Harofteh B, Etemadifar M (2022) Autoimmune meningoencephalitis associated with anti-glutamic acid decarboxylase antibody following COVID-19 infection: A case report. Clin Case Rep 10, e6597.
[26] Stadelman AM, Ssebambulidde K, Buller A, Tugume L, Yuquimpo K, Bakker CJ, Boulware DR, Bahr NC (2022) Cerebrospinal fluid AFB smear in adults with tuberculous meningitis: A systematic review and diagnostic test accuracy meta-analysis. Tuberculosis (Edinb) 135, 102230.
[27] Seddon JA, Tugume L, Solomons R, Prasad K, Bahr NC, Tuberculous Meningitis International Research Consortium (2019) The current global situation for tuberculous meningitis: epidemiology, diagnostics, treatment and outcomes. Wellcome Open Res 4, 167.
[28] Mawanda F, Wallace R (2013) Can infections cause Alzheimer's disease? Epidemiol Rev 35, 161-180.
[29] Broxmeyer L (2017) Dr. Oskar Fischer’s Mysterious Little Alzheimer’s Germ. J Alzheimers Dis Editor’s blog. https://www.j-alz.com/editors-blog/posts/dr-oskar-fischers-mysterious-li...
[30] Tomiyama T, Asano S, Suwa Y, Morita T, Kataoka K, Mori H, Endo N (1994) Rifampicin prevents the aggregation and neurotoxicity of amyloid beta protein in vitro. Biochem Biophys Res Commun 204, 76-83.
[31] Umeda T, Ono K, Sakai A, Yamashita M, Mizuguchi M, Klein WL, Yamada M, Mori H, Tomiyama T (2016) Rifampicin is a candidate preventive medicine against amyloid-β and tau oligomers. Brain 139, 1568–1586.
[32] Iizuka T, Morimoto K, Sasaki Y, Kameyama M, Kurashima A, Hayasaka K, Ogata H, Goto H (2017) Preventive effect of rifampicin on Alzheimer disease needs at least 450 mg daily for 1 year: an FDG-PET follow-up study. Dement Geriatr Cogn Disord Extra 7, 204-214.
[33] Sethi NK, Sethi PK, Torgovnick J, Arsura E (2011) Central nervous system tuberculosis masquerading as primary dementia: a case report. Neurol Neurochir Pol 45, 510-513.
[34] Henkle E, Winthrop KL, Ranches GP, et al. (2020) Preliminary validation of the NTM Module: a patient-reported outcome measure for patients with pulmonary nontuberculous mycobacterial disease. Eur Respir J 55, 1901300.
[35] Kawka M, Brzostek A, Dzitko K, Kryczka J, Bednarek R, Płocińska R, Płociński P, Strapagiel D, Gatkowska J, Dziadek J, Dziadek B (2021) Mycobacterium tuberculosis binds human serum amyloid A, and the interaction modulates the colonization of human macrophages and the transcriptional response of the pathogen. Cells 10, 1264.
[36] De Beer FC, Ne AE, Gie RP, Donald PR, Stracham AF (1984) Serum amyloid A protein and C-reactive protein levels in pulmonary tuberculosis: Relationship to amyloidosis. Thorax 39, 196–200.
[37] De Groote MA, Sterling DG, Hraha T, Russell TM, Green LS, Wall K, Kraemer S, Ostroff R, Janjic N, Ochsner UA (2017) Discovery and validation of a six-marker serum protein signature for the diagnosis of active pulmonary tuberculosis. J Clin Immunol 55, 3057-3071.
[38] Jiang TT, Shi LY, Wei LL, Li X, Yang S, Wang C, Liu CM, Chen ZL, Tu HH, Li ZJ, Li JC (2017) Serum amyloid A, protein Z, and C4b-binding protein β chain as new potential biomarkers for pulmonary tuberculosis. PLoS One 2017, 0173304.
[39] Schwartz P (1970) Amyloidosis—Cause and Manifestation of Senile Deterioration. Charles C Thomas, Springfield, IL, 363 pp.
[40] Wang L, Maji SK, Sawaya MR, Eisenberg D, Riek R (2008) Bacterial inclusion bodies contain amyloid-like structure. PLoS Biol 6, e195.
[41] Hsu T, Hingley-Wilson SM, Chen B, Chen M, Dai AZ, Morin PM, Marks CB, Padiyar J, Goulding C, Gingery M, Eisenberg D, Russell RG, Derrick SC, Collins FM, Morris SL, King CH, Jacobs WR Jr. (2003) The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A 100, 12420-12425.
[42] Lansbury PT Jr. (1999) Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci U S A 96, 3342-3344.
[43] Belyaeva IV, Kosova AN, Vasiliev AG (2022) Tuberculosis and autoimmunity. Pathophysiology 29, 298-318. Erratum in: Pathophysiology. 2022 Aug 16;29(3):469-470.
[44] Broxmeyer L (2016) Alzheimer's disease–How Its Bacterial Cause Was Found and Then Discarded. CreateSpace Independent Publishing Platform. 190 pp.







